基于GCN的分子性质预测
前文讲到图数据天然适合描述分子的二维结构数据,本文将使用ECOL数据集实现一个分子性质预测模型,以下是代码详解。
项目目录
本项目的目录结构如下:
GCN Molecular Property/
├── data/ # 数据集文件
│ └── ECOL.csv
├── result/ # 结果目录
│ ├── model.pth # 模型权重
│ └── losses.csv # 平均损失记录
├── ChemGraph.py # 化学结构转换工具
├── config.py # 超参数配置文件
├── dataset.py # 加载数据集
├── GCNModel.py # GCN模块
├── predict.py # 推理预测代码
└── train.py # 训练代码超参数配置
这里主要设置了数据集文件路径和模型权重保存路径,批次大小、训练轮数、学习率等。其中最主要的是节点向量维度N_DIM,该值必须和graph中的节点向量维度一致,在不同的学习任务中可能会选择不同的原子节点属性,这会造成节点维度的变化,需要特别注意输入参数的一致性。
以下代码在config.py中实现:
import torch
import torch.nn as nn
# 设备
DEVICE = "cuda" if torch.cuda.is_available() else "cpu"
# 数据集路径
DELANEY_FILE = "./data/ECOL.csv"
# 权重保存文件
PT_PATH = "./result/model.pth"
# 损失保存路径
LOSS_PATH = "./result/losses.csv"
# 节点特征向量维度
N_DIM = 22
# 批次大小
BATCH_SIZE = 32
# 训练轮数
EPOCH = 200
# 学习率
LR = 5e-3化学结构工具
以下代码在ChemGraph.py中实现:
为了将化学结构转变为计算机可以理解的数据,需要借助RDkit库和PyG库,将mol对象变为图结构,构建的图数据包含节点特征、边索引、边特征三个张量。
from rdkit import Chem
from rdkit.Chem import rdchem
import numpy as np
import torch
from torch_geometric.data import Data
import networkx as nx
import matplotlib.pyplot as plt
ATOM_TYPE = ('C', 'O', 'N','H', 'P', 'S', 'F', 'Cl', 'Br', 'I', 'UNK')
ATOM_LEN = len(ATOM_TYPE)
HYBRIDS = (
Chem.rdchem.HybridizationType.SP,
Chem.rdchem.HybridizationType.SP2,
Chem.rdchem.HybridizationType.SP3,
Chem.rdchem.HybridizationType.SP3D,
Chem.rdchem.HybridizationType.SP3D2,
'UNK'
)
HYB_LEN = len(HYBRIDS)
BIND_TYPE = (
rdchem.BondType.SINGLE,
rdchem.BondType.DOUBLE,
rdchem.BondType.TRIPLE,
rdchem.BondType.AROMATIC
)
BIND_LEN = len(BIND_TYPE)接受smiles式输出图数据
# 从分子生成图
def mol_to_graph(mol: rdchem.Mol) -> Data:
nodes_features = []
for atom in mol.GetAtoms():
node = []
# 原子类型编码
atom_hot = [0] * ATOM_LEN
atom_symbol = atom.GetSymbol() if atom.GetSymbol() in ATOM_TYPE else 'UNK'
idx = ATOM_TYPE.index(atom_symbol)
atom_hot[idx] = 1
# 原子杂化编码
hyb_hot = [0] * HYB_LEN
hyb = atom.GetHybridization() if atom.GetHybridization() in HYBRIDS else "UNK"
idx = HYBRIDS.index(hyb)
hyb_hot[idx] = 1
other_feats = [
float(atom.GetIsAromatic()), # 芳香族(布尔转0/1)
atom.GetTotalValence(), # 总价电子数
atom.GetDegree(), # 配位数
atom.GetFormalCharge(), # 形式电荷
atom.GetNumRadicalElectrons() # 自由基电子数
]
node = atom_hot+hyb_hot+other_feats
nodes_features.append([float(f) for f in node])
x = torch.tensor(nodes_features, dtype=torch.float)
# 边索引和边特征
edge_indices = []
edge_features = []
for bond in mol.GetBonds():
u, v = bond.GetBeginAtomIdx(), bond.GetEndAtomIdx()
# 无向图双向边
edge_indices.extend([[u, v], [v, u]])
bond_type = bond.GetBondType()
# 独热编码键类型:[单键, 双键, 三键, 芳香键]
bind_hot = [0] * BIND_LEN
idx = BIND_TYPE.index(bond_type)
bind_hot[idx] = 1
# 双向边特征相同
edge_features.extend([bind_hot,bind_hot])
edge_index = torch.tensor(edge_indices, dtype=torch.long).t().contiguous()
edge_attr = torch.tensor(edge_features, dtype=torch.float)
return Data(
x=x, # 节点特征
edge_index=edge_index, # 边索引
edge_attr=edge_attr, # 边特征
num_nodes=mol.GetNumAtoms() # 节点数
)从图数据还原为mol对象
# 从图还原分子
def graph_to_mol(graph: Data) -> rdchem.Mol:
# 初始化分子
mol = Chem.RWMol()
# 遍历节点
for node in graph.x.numpy():
# 前 ATOM_LEN 为原子序数
atom_type_idx = np.argmax(node[:ATOM_LEN])
atom_symbol = ATOM_TYPE[atom_type_idx]
if atom_symbol == 'UNK':
# 未知原子默认用碳替代
atom_symbol = 'C'
atom = Chem.Atom(atom_symbol)
# 杂化类型
hyb_start = ATOM_LEN
hyb_end = hyb_start + HYB_LEN
hyb_idx = np.argmax(node[hyb_start:hyb_end])
hybrid_type = HYBRIDS[hyb_idx]
if hybrid_type != 'UNK':
atom.SetHybridization(hybrid_type)
# 解析芳香性
is_aromatic = bool(round(node[hyb_end]))
atom.SetIsAromatic(is_aromatic)
# 添加原子到分子
mol.AddAtom(atom)
edges_index = graph.edge_index.T.numpy()
edges_type = graph.edge_attr.numpy()
for i in range(len(edges_index)//2):
u, v = edges_index[2*i][0], edges_index[2*i][1]
bind_type = BIND_TYPE[np.argmax(edges_type[2*i])]
mol.AddBond(int(u), int(v), bind_type)
try:
# 校验并修复分子(如价态、隐式氢)
Chem.SanitizeMol(mol)
mol.UpdatePropertyCache(strict=False)
except Exception as e:
print(f"分子修正警告:{e},返回未修正分子")
return mol可视化展示生成的图数据,同时显示节点元素符号、连接状态和边特征(成键类型)
# 分子图可视化函数
def visualize_molecule(pyg_graph, figsize=(9, 7)):
# 构建NetworkX图并提取唯一边(去重双向边)
G = nx.Graph()
G.add_nodes_from(range(pyg_graph.num_nodes))
edges = list(zip(pyg_graph.edge_index[0].numpy(), pyg_graph.edge_index[1].numpy()))
unique_edges = []
seen = set()
for u, v in edges:
if (u, v) not in seen and (v, u) not in seen:
seen.add((u, v))
unique_edges.append((u, v))
G.add_edges_from(unique_edges)
# 节点配置:原子序数作为标签,颜色/大小区分原子类型
node_labels = {}
node_colors = []
for i, node in enumerate(pyg_graph.x.numpy()):
index = np.argmax(node[:ATOM_LEN])
atom_symbol = ATOM_TYPE[index]
node_labels[i] = atom_symbol
atom_number = Chem.Atom(atom_symbol).GetAtomicNum()
node_colors.append(atom_number)
# 边配置:从edge_attr(独热编码)解析键类型
bond_symbols = ['-', '=', '≡', '*']
edge_labels = {}
for i, (u, v) in enumerate(unique_edges):
bond_feat = pyg_graph.edge_attr[i * 2].numpy()
bond_idx = bond_feat.argmax()
edge_labels[(u, v)] = bond_symbols[bond_idx]
# 布局选择:根据分子结构自动适配
# 判断是否含环(简单规则:存在节点度数≥3)
has_ring = any(G.degree(node) >= 3 for node in G.nodes)
if has_ring:
pos = nx.kamada_kawai_layout(G, scale=3)
else:
pos = nx.spring_layout(G, seed=42, k=2.0, iterations=100)
# 绘制图形
fig, ax = plt.subplots(figsize=figsize)
# 绘制边
nx.draw_networkx_edges(
G, pos, ax=ax,
edge_color='#555555', width=2, alpha=0.8
)
# 绘制节点
scatter = nx.draw_networkx_nodes(
G, pos, ax=ax,
node_color=node_colors,
cmap=plt.cm.Pastel1, edgecolors='#222222', linewidths=1.5
)
# 绘制节点标签(原子序数)
nx.draw_networkx_labels(
G, pos, ax=ax, labels=node_labels,
font_size=11, font_weight='bold',
)
# 绘制边标签(键类型)
nx.draw_networkx_edge_labels(
G, pos, ax=ax, edge_labels=edge_labels,
font_size=13, font_color='#D62828',
bbox=dict(boxstyle='round,pad=0.2', facecolor='white', alpha=0.8)
)
# 美化设置
ax.set_title('Molecular Graph Visualization', fontsize=14, fontweight='bold', pad=20)
ax.axis('off') # 隐藏坐标轴
plt.tight_layout()
plt.show()最后编写测试模块,将阿司匹林分子smiles式转为图,并显示图结构。
if __name__ == '__main__':
smiles = "CC(=O)OC1=CC=CC=C1C(=O)O"
mol = Chem.MolFromSmiles(smiles)
G = mol_to_graph(mol)
visualize_molecule(G)
new_smiles = graph_to_mol(G)
print("new smiles:", Chem.MolToSmiles(new_smiles))下图展示的阿司匹林分子图可视化,观察边属性和节点属性和分子信息一致: 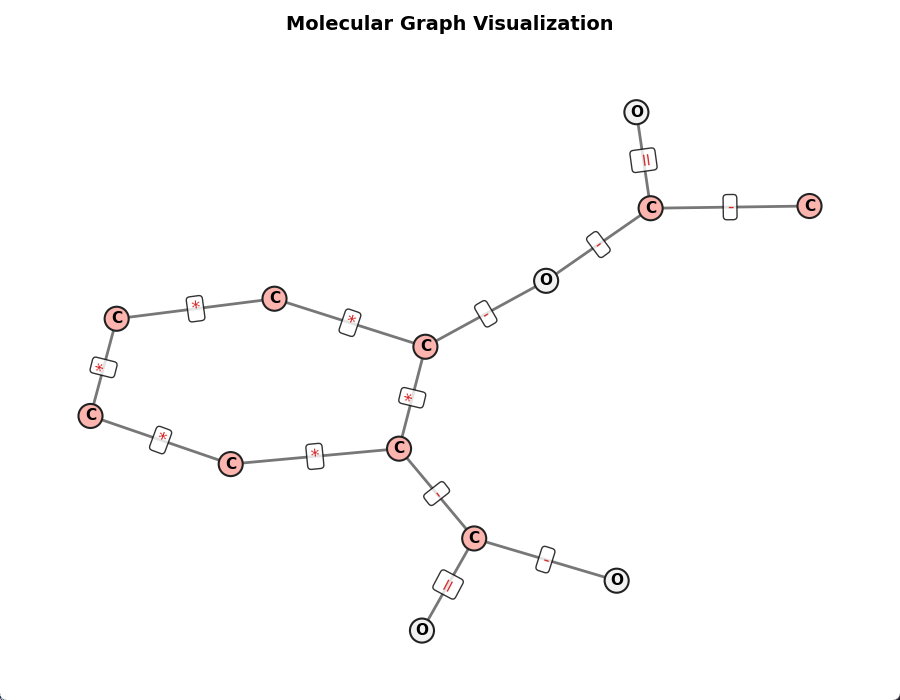
加载数据集
以下代码在dataset.py中实现:
需要导入csv文件中的数据,生成mol对象,选择需要的数据,构造新的df对象。
import pandas as pd
from rdkit import Chem
import torch
from torch.utils.data import Subset
from torch_geometric.loader import DataLoader
from torch_geometric.data import Dataset
from ChemGraph import mol_to_graph
from config import *
# 选择要学习预测的指标
TASK = 'ESOL predicted log solubility in mols per litre'
# 读取数据集csv文件
df = pd.read_csv(DELANEY_FILE)
# 将smiles转为rdkit的mol对象
df["mol"] = df["smiles"].apply(lambda x: Chem.MolFromSmiles(x))
# 去除转化失败的空值
df = df[df["mol"].notna()]
# 选择属性构造新列表
df = df[["Compound ID", "smiles", TASK,"mol"]]
# 预计算所有graph
if "graph" not in df.columns:
df["graph"] = df["mol"].apply(lambda x: mol_to_graph(x))
# 过滤转换失败的样本(若有)
df = df[df["graph"].notna()].reset_index(drop=True)定义MolGraphDataset,继承Data类,并实现get方法。加载后的数据集应该包含节点特征向量、边索引张量、边属性张量、化学性质张量。
# 数据类,继承Dataset
class MolGraphDataset(Dataset):
# 初始化方法需传入df对象、mol对象列名、选择的属性、转换函数
def __init__(self, df, # df对象
mol_col="mol", # mol对象所在列名
target_col=TASK, # 选择要学习的属性列
transform_fn=None # 预处理函数
):
super().__init__()
self.df = df
self.mol_col = mol_col
self.target_col = target_col
self.transform_fn = transform_fn
# len方法
def len(self):
return len(self.df)
# get方法
def get(self, idx):
row = self.df.iloc[idx]
mol = row[self.mol_col]
try:
G = row["graph"]
# 在graph中增加要学习的性质张量
if self.target_col is not None:
target = torch.tensor([row[self.target_col]], dtype=torch.float)
G.y = target
return G
except:
return None随后实例化数据类,设置划分比例和随机种子,将数据集随即划分为训练集/测试集,生成DataLoader可迭代对象。
# 初始化完整的分子图数据集
full_dataset = MolGraphDataset(
df=df,
mol_col="mol",
target_col=TASK,
)
data_size = len(full_dataset)
# 划分比例
test_ratio = 0.10
# 测试集大小
test_size = int(data_size * test_ratio)
# 训练集大小
train_size = data_size - test_size
# 生成随机打乱的索引
random_seed = 42
torch.manual_seed(random_seed)
indices = torch.randperm(data_size)
# 分割为训练集和测试集索引
train_indices = indices[:train_size]
test_indices = indices[train_size:]
# 用Subset构建训练集和测试集
train_dataset = Subset(full_dataset, train_indices)
test_dataset = Subset(full_dataset, test_indices)
# 构建数据加载器
train_loader = DataLoader(
train_dataset,
batch_size=BATCH_SIZE,
shuffle=True, # 训练集打乱
drop_last=False, # 不丢弃最后一个不足批次的数据
num_workers=0, # 多进程加载
pin_memory=True # 加速传输
)
test_loader = DataLoader(
test_dataset,
batch_size=BATCH_SIZE,
shuffle=False, # 测试集不打乱
drop_last=False,
num_workers=0,
pin_memory=True
)经过DataLoader打包后,节点特征x会被纵向拼接[总节点数, 节点特征维度] ;
边索引 edge_index 会被调整偏移量后拼接[2,总边数],确保不同图的边索引不冲突;
边属性 edge_attr 会变成[总边数,边特征维度];
每个图的标签 y 会被保存在 Batch.y 中[批大小, 标签维度]。
单元模块测试,输出数据集的基本信息。
if __name__ == '__main__':
print(f"Number of molecules in the dataset: {df.shape[0]}")
print(df.head())
print(f"完整数据集大小: {data_size}")
print(f"训练集大小: {len(train_dataset)}, 测试集大小: {len(test_dataset)}")
print("验证训练集批次:")
for batch in train_loader:
print(f"节点特征形状: {batch.x.shape}")
print(f"标签形状: {batch.y.shape}")
print(f"边索引形状: {batch.edge_index.shape}")
print(f"边属性形状: {batch.edge_attr.shape}")
break输出内容如下:
Number of molecules in the dataset: 1128
Compound ID SMILES logS mol graph
0 Amigdalin OCC3OC(OCC2OC(OC(C#N)c1ccccc1)C(O)C(O)C2O)C(O)... -0.974 <rdkit.Chem.rdchem.Mol object at 0x00000217ADB... [(x, [tensor([0., 1., 0., 0., 0., 0., 0., 0., ...
1 Fenfuram Cc1occc1C(=O)Nc2ccccc2 -2.885 <rdkit.Chem.rdchem.Mol object at 0x00000217ADB... [(x, [tensor([1., 0., 0., 0., 0., 0., 0., 0., ...
2 citral CC(C)=CCCC(C)=CC(=O) -2.579 <rdkit.Chem.rdchem.Mol object at 0x00000217ADB... [(x, [tensor([1., 0., 0., 0., 0., 0., 0., 0., ...
3 Picene c1ccc2c(c1)ccc3c2ccc4c5ccccc5ccc43 -6.618 <rdkit.Chem.rdchem.Mol object at 0x00000217ADB... [(x, [tensor([1., 0., 0., 0., 0., 0., 0., 0., ...
4 Thiophene c1ccsc1 -2.232 <rdkit.Chem.rdchem.Mol object at 0x00000217ADB... [(x, [tensor([1., 0., 0., 0., 0., 0., 0., 0., ...
完整数据集大小: 1128
训练集大小: 1016, 测试集大小: 112
验证训练集批次:
节点特征形状: torch.Size([833, 22])
标签形状: torch.Size([64])
边索引形状: torch.Size([2, 1740])
边属性形状: torch.Size([1740, 4])模型定义
以下代码在GCNModel.py中实现:
利用GCN图卷积模块构建预测网络,为了快速入门测试,这个简单的GCN模块没有融合边的特征。
import torch.nn as nn
from torch_geometric.nn.conv import GCNConv
from torch_geometric.nn.pool import global_mean_pool
class GCNModel(nn.Module):
def __init__(self, ndim, hidden_dims, dropout=0.2):
super(GCNModel, self).__init__()
dims = [ndim] + hidden_dims
# 存储所有GCNConv层
self.convs = nn.ModuleList()
# 存储所有激活层
self.acts = nn.ModuleList()
# 存储所有Dropout层
self.dropouts = nn.ModuleList()
# 归一化层
self.bn = nn.BatchNorm1d(dims[0])
for i in range(len(dims)-1):
self.convs.append(GCNConv(dims[i], dims[i+1]))
self.acts.append(nn.ReLU())
self.dropouts.append(nn.Dropout(dropout))
# 线性层
self.fc = nn.Linear(dims[-1], 1)
def forward(self, data):
out = self.bn(data.x)
edge_index = data.edge_index.long()
# 逐层调用,显式传递edge_index给GCNConv
for conv, act, dropout in zip(self.convs, self.acts, self.dropouts):
# GCNConv层
out = conv(out, edge_index)
# ReLU层
out = act(out)
# Dropout层
out = dropout(out)
out = global_mean_pool(out, data.batch)
return self.fc(out)测试模块
if __name__ == '__main__':
from config import *
model = GCNModel(ndim=N_DIM, hidden_dims=[128, 64, 32])
model = model.to(DEVICE)
model = model.float()
optimizer = torch.optim.Adam(model.parameters(), lr=LR)
criterion = nn.MSELoss()
print("Number of trainable parameters:",
sum(p.numel() for p in model.parameters() if p.requires_grad))
print(model)打印的模型结构信息:
Number of trainable parameters: 13357
GCNModel(
(bn): BatchNorm1d(22, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(net): Sequential(
(0): GCNConv(22, 128)
(1): ReLU()
(2): GCNConv(128, 64)
(3): ReLU()
(4): GCNConv(64, 32)
(5): ReLU()
)
(fc): Linear(in_features=32, out_features=1, bias=True)
)训练模型
以下代码在train.py中实现:
import numpy as np
import matplotlib.pyplot as plt
from tqdm import tqdm
import torch
import torch.nn as nn
from dataset import *
from ChemGraph import *
from GCNModel import *
from config import *
# 训练周期
def train_epoch(model, criterion, optimizer, dataloader):
losses = []
# 训练模式
model.train()
for G in dataloader:
G = G.to(DEVICE, non_blocking=True)
# 获取属性标签
y_true = G.y
# 清空梯度
optimizer.zero_grad()
# 生成模型预测值
y_pred = model(G)
# 计算损失函数
loss = criterion(y_pred, y_true.reshape(y_pred.shape))
# 反向传播
loss.backward()
# 更新权重
optimizer.step()
# 记录损失值
losses.append(loss.cpu().detach().item())
return losses
# 测试周期
def val_epoch(model, criterion, dataloader):
losses = []
# 评估模式
model.eval()
# 关闭梯度更新
with torch.no_grad():
for G in dataloader:
G = G.to(DEVICE, non_blocking=True)
# 真实值
y_true = G.y
# 模型预测值
y_pred = model(G)
# 计算误差
loss = criterion(y_pred, y_true.reshape(y_pred.shape))
# 记录损失
losses.append(loss.cpu().detach().item())
return losses正式训练部分,模型的训练和评估同时进行:
if __name__ == '__main__':
# 模型初始化
model = GCNModel(ndim=N_DIM, hidden_dims=[128, 64, 32])
# 迁移设备
model.to(DEVICE)
# 确保模型参数为float类型
model = model.float()
# 优化器
optimizer = torch.optim.Adam(model.parameters(), lr=LR)
# 均方差损失函数
criterion = nn.MSELoss()
# 训练误差
train_loss = []
# 评估误差
val_loss = []
# 初始化进度条
loss_dict = {"avg_train_loss":0,"avg_val_loss":0}
pbar = tqdm(
range(1, EPOCH+1),
desc="Epoch",
leave=True,
postfix=loss_dict,
dynamic_ncols=True
)
# 训练循环
for epoch in pbar:
# 训练并更新权重
losses = train_epoch(model, criterion, optimizer, train_loader)
avg_train_loss = np.mean(losses)
train_loss.append(avg_train_loss)
# 评估
losses = val_epoch(model, criterion, test_loader)
avg_val_loss = np.mean(losses)
val_loss.append(avg_val_loss)
# 更新进度条参数
loss_dict["avg_train_loss"] = avg_train_loss
loss_dict["avg_val_loss"] = avg_val_loss
pbar.set_postfix(loss_dict)
# 保存模型
torch.save(model.state_dict(), PT_PATH)
print("\n训练完成!模型权重已保存为:", PT_PATH)
# 保存每轮损失
import pandas as pd
df = pd.DataFrame({
'Epoch': range(1, len(train_loss) + 1),
'Training Loss': train_loss,
'Test Loss': val_loss
})
df.to_csv(LOSS_PATH, index=False)
# 展示损失函数
f, ax = plt.subplots(1, 1, figsize=(5,5))
ax.plot(train_loss, c="blue", label="Training")
ax.plot(val_loss, c="red", label="Test")
plt.xlabel("Epoch")
plt.ylabel("Loss")
plt.legend()
plt.show()整个训练过程的输出如下:
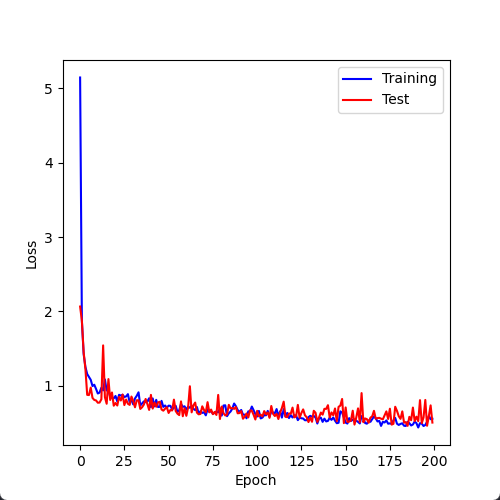
Epoch: 100%|██████| 200/200 [00:30<00:00, 6.58it/s, avg_train_loss=0.542, avg_val_loss=0.508]
训练完成!模型权重已保存为: ./result/model.pth采样两种模型训练后的全程平均误差曲线图:
预测模型
最后实现一个单输入预测函数,利用训练好的模型对给定smiles分子进行性质预测。
以下代码实现在predict.py中:
from rdkit import Chem
import torch
from ChemGraph import *
from config import *
from GCNModel import *
# 单输入预测
def predict(smiles, model, transform_fn, device):
# 将SMILES转换为RDKit的Mol对象
mol = Chem.MolFromSmiles(smiles)
if mol is None:
print(f"SMILES {smiles} 无效,无法转换为分子!")
return None
# 转换为PyG的Data对象(同训练时的处理)
data = transform_fn(mol)
data = data.to(device)
# 前向传播预测
with torch.no_grad():
prediction = model(data)
# 返回预测值(转换为numpy或Python数值)
return prediction.cpu().numpy()[0, 0]
if __name__ == '__main__':
model = GCNModel(ndim=N_DIM, hidden_dims=[128, 64, 32])
model = model.to(DEVICE)
model = model.float()
model.load_state_dict(torch.load(PT_PATH, map_location=DEVICE))
model.eval()
smiles = "COC"
prediction = predict(
smiles=smiles,
model=model,
transform_fn=mol_to_graph,
device=DEVICE
)
print(f"分子 {smiles} ;预测性质值为: {prediction:.4f}")模型的输出值:
分子 COC ;预测性质值为: 0.2920